国际肝病 发表时间:2026/6/28 20:37:10 浏览量:147
编者按:遗传代谢性肝病是一组由基因突变引发的肝脏疾病,并可能伴发多器官、多系统的损害,多发生于儿童或青少年,也可见于成人。此类疾病常由于起病隐匿、临床表现不具特异性,且多种病因可以表现出类似症状,早期诊断极具挑战性。为提高诊断效率和准确性,采取以临床思维为主导的综合性评估策略显得尤为重要。在“2026年世界华人医师协会肝脏病学专业委员会病理专题会暨第十届病理大师班”上,首都医科大学附属北京佑安医院病理科刘晖教授在主题报告中,系统阐述了遗传代谢性肝病的病理诊断思路,依据组织学特征将其分为肝组织基本正常型、脂肪变性型、胆汁淤积型、贮积/沉积型、肝炎型和门静脉高压型六大类型,并结合典型病例展示了各类型的病理特点与鉴别诊断要点,为临床病理诊断提供了系统指导。
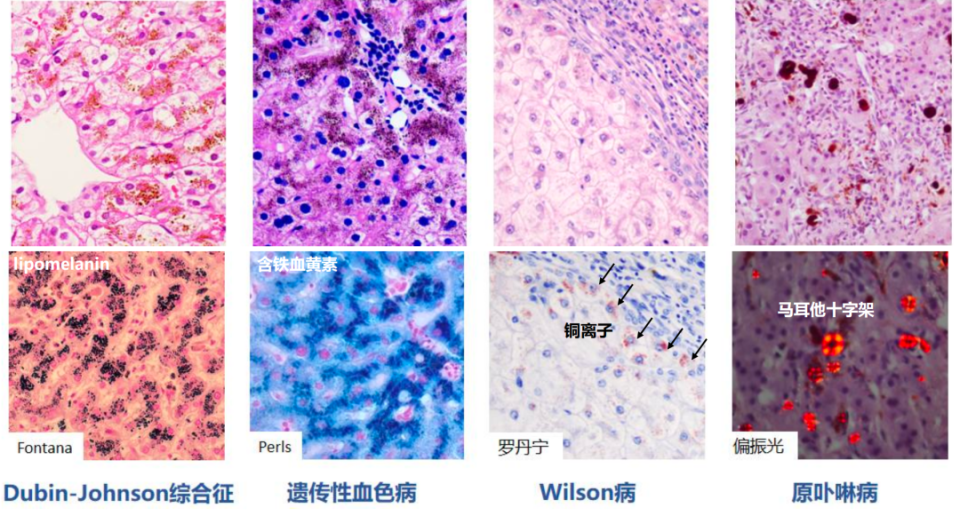
遗传代谢性肝病的病理诊断需结合光镜、电镜、特殊染色及免疫组化等多种技术手段。部分疾病在光镜下具有特征性组织学表现,如原卟啉病、Dubin-Johnson综合征、胆管板发育畸形,一些贮积类疾病,如尼曼-匹克病、戈谢病、胆固醇酯贮积病、a1-抗胰蛋白酶缺乏症等,在光镜下也有特征表现。有的疾病通过电镜观察特征性超微结构,如α1-抗胰蛋白酶缺乏症、Farber病、糖原贮积症(Ⅱ/Ⅳ型)、遗传性果糖不耐受、戈谢病、异染性脑白质营养不良、神经节苷脂沉积症、Dubin-Johnson综合征、Wilson病、Zellweger综合征、婴儿型Refsum病等;对于无特征性组织学表现的疾病,则需结合特殊染色、免疫组化及临床表现缩小鉴别诊断范围。
根据组织学形态特征,遗传代谢性肝病可分为六大类型:肝组织基本正常型、脂肪变性型、胆汁淤积型、贮积/沉积型、肝炎型和门脉高压型[1]。各型可单独出现,多数可合并存在。
少数肝脏代谢性疾病表现为肝功能异常,组织学改变轻微或无变化,多不引起持续肝损伤。肝组织基本正常型主要包括Gilbert综合征、Roter综合征、苯丙酮尿症、胱氨酸血症、尿素循环障碍、氨基酸代谢障碍等。这类疾病肝组织学无明显异常,诊断主要依赖临床表现和基因检测。
图1. Gilbert综合征、Roter综合征的组织学特征
Gilbert综合征和Roter综合征是我们较为熟知的两种疾病,肝脏组织学表现基本正常。另外介绍一个疾病,我们观察到它的组织学也是基本正常或有轻微改变,即遗传性高胆汁酸血症——钠-牛磺胆酸共转运多肽(NTCP)缺陷病(SLC10A1基因突变)也属于此型。
典型病例:女性,30岁,间断总胆汁酸升高12年。肝功能示ALT 13 U/L,AST 20 U/L,ALP 59 U/L,GGT 13 U/L,TBIL 19.2 μmol/L,DBIL 6.2 μmol/L,ALB 46.2 g/L,TBA 82.8 μmol/L。基因检测示SLC10A1基因exon4 c.800C>T p.S267F纯合突变。该病为常染色体隐性遗传,表现为胆汁酸水平升高。
肝组织学所见主要为肝细胞脂肪变性,除单纯性脂肪肝外,应考虑存在以下疾病的可能:Wilson病、糖原贮积症(Ⅰ、Ⅲ型)、囊性纤维化(CF)、半乳糖血症、果糖醛缩酶缺乏(果糖不耐受)、肉碱酰基转移酶缺乏、尿素循环障碍、Citrin缺乏症、脂肪酸氧化缺陷、线粒体病、氨基酸代谢病等。
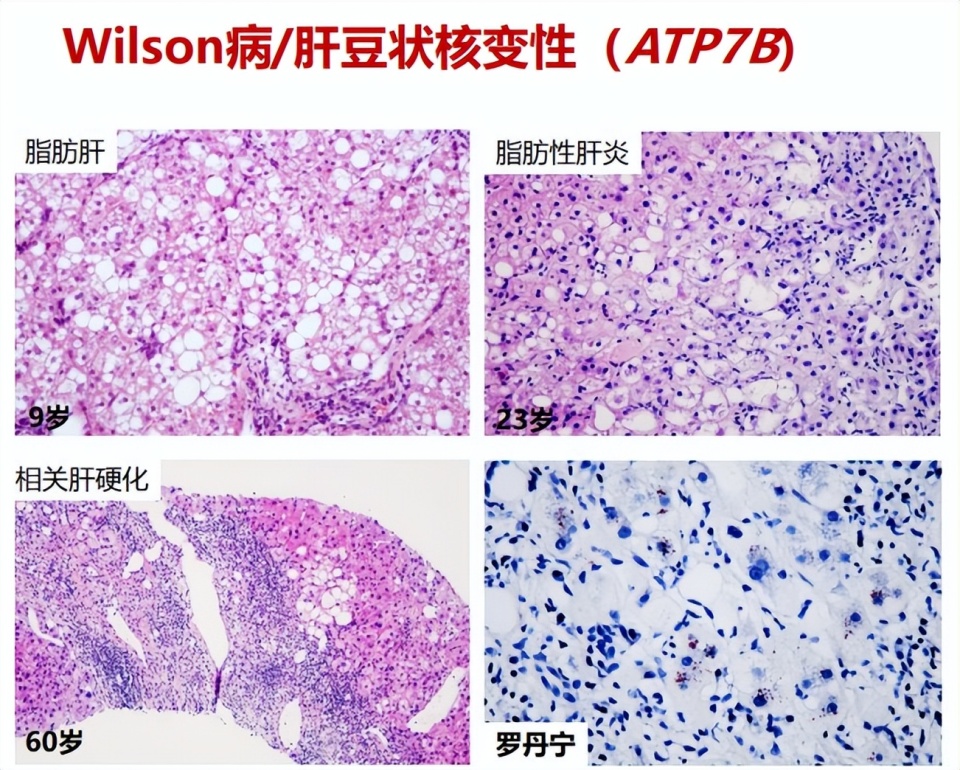
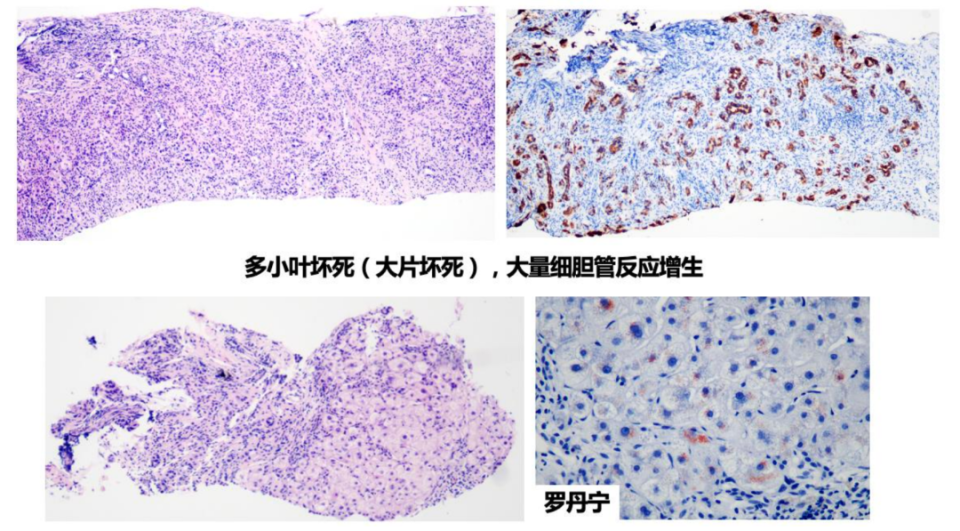
Wilson病是遗传代谢性肝病中组织学表现最为多样的疾病,且无组织学特异性,可表现为脂肪性肝炎、急性肝炎、暴发性肝炎、慢性肝炎、肝硬化,其中脂肪变性可出现在各阶段中,是早期及中期最主要的表现。应用Rhodanine/Timms染色,如发现肝组织中过多铜颗粒沉积,在排除胆汁淤积性肝病后,常提示Wilson病诊断。
图2. Wilson病各阶段的组织学特征
SLC25A13基因突变导致线粒体天冬氨酸-谷氨酸载体蛋白Citrin功能不足,婴儿表现为新生儿肝内胆汁淤积;成人起病的Citrin缺乏症常出现高氨血症,喜高蛋白饮食,厌碳水化合物食物,组织学呈肝细胞大泡性脂肪变性、窦周纤维化。
典型病例:男性,44岁。喜食高蛋白食物,厌食甜食,有神经精神症状,间断发作,多次住院,高氨血症。血氨基酸检测示瓜氨酸230.927μmol/L(正常7-18.7),谷氨酸139.028μmol/L(正常38-98),鸟氨酸150.293μmol/L(正常17-49.6),丝氨酸101.062μmol/L(正常26-67)。
溶酶体酸性脂肪酶缺乏症(LALD)是由于LIPA基因突变使溶酶体酸性脂肪酶缺乏,导致胆固醇酯和三酰甘油在肝、脾、肾上腺及心血管系统等组织贮积,为罕见的常染色体隐性遗传病。根据发病年龄和临床表现不同,分为婴儿期起病的Wolman病和儿童及成人期起病的胆固醇酯贮积病(CESD)。
儿童及成人型病例:1)8岁女性,BMI 21.1,肝细胞内大小泡混合型脂变,汇管区轻度纤维化,初诊为NAFLD;3年后,电镜显示肝细胞及Kupffer细胞内微泡脂变及脂肪囊泡。2)16岁女性,BMI 53.18,肝细胞内大小泡混合型脂变,纤维化3-4期,电镜显示肝细胞内脂肪囊泡。
包含多种类型,形态不尽相同,除Ⅱ型外,其余类型均为非溶酶体贮积,表现为胞浆内的贮积。大部分糖原贮积症组织学突出的特点是肝细胞肿胀含丰富糖原,胞质淡染、苍白,细胞膜清晰,细胞核固缩、位于中心或偏位,同时可见明显肝细胞脂肪变性。
典型病例:5岁女性,间断上腹痛20余天,CT示肝大、不均质脂肪肝。除脂变外,尚有核膜清晰胞浆淡染的细胞,PAS染色呈紫红色,基因检测结果显示糖原贮积症6型。
以小泡性脂变为主,胞浆内小脂滴均匀分布,围绕肝细胞核,大泡性脂变也可共存。MVP17基因突变所致的线粒体耗竭综合征表现为肝细胞变性、坏死、脂变。
胆汁淤积型主要包括家族性肝内胆汁淤积和先天性胆管发育不良等。
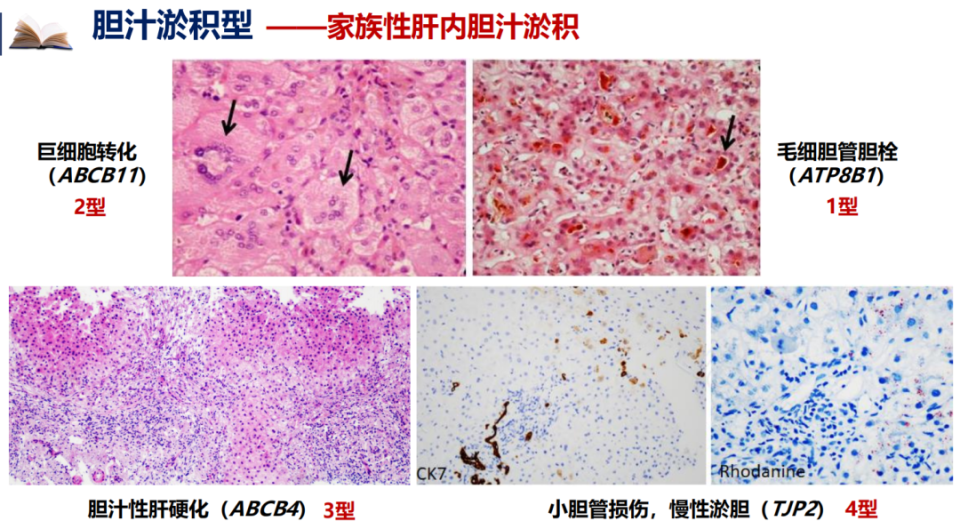
家族性肝内胆汁淤积症(FIC)是一组编码与胆汁流形成和稳态相关分子的基因发生变异导致的,以肝内胆汁淤积为主要表现的常染色体隐性遗传病。根据血γ-谷氨酰转移酶(GGT)水平是否增高,可分为低GGT型和高GGT型。严重者通常在婴幼儿期起病,表现为持续性胆汁淤积,可进展为终末期肝病,称为进行性家族性肝内胆汁淤积症(PFIC),是我国儿童慢性肝病和儿童肝移植的重要原因[2]。
FIC根据致病基因不同可分为多种亚型。巨细胞转化见于ABCB11突变(2型),毛细胆管胆栓见于ATP8B1突变(1型),胆汁性肝硬化见于ABCB4突变(3型),以上3种类型是我们较为熟知的,TJP2突变所致的FIC4型,表现为小胆管损伤、慢性胆汁淤积。
图3. FIC不同亚型的组织学特征
病例:男性,14岁,发现皮肤巩膜黄染、尿黄1个月。肝功能示ALT 60 U/L,AST 87 U/L,TBIL 178.8μmol/L,DBIL 77.5μmol/L,ALP 495 U/L,GGT 188.5 U/L。肝穿刺活检示胆汁性肝硬化,毛细胆管内可见胆栓。基因检测示HSD3B7基因复合杂合突变(c.171_172del p.P57fs和c.682C>T p.R228W)。HSD3B7编码3β-羟基-Δ5-C27-类固醇脱氢酶/异构酶,是参与胆固醇合成胆汁酸途径的关键酶,基因突变导致胆汁酸合成中断,毒性中间产物堆积,导致肝损伤,属于先天性胆汁酸合成障碍。
先天性肝内胆管缺乏也可导致胆汁淤积,代表性疾病是Alagille综合征,是一种累及多系统的常染色体显性遗传疾病。Alagille综合征主要由JAG1或Notch2基因变异导致Notch信号通路异常所致,其中约94.3%的患者存在编码Jagged1的JAG1基因(20p12)突变,符合经典临床特征,称之为Alagille综合征1型;约2.5%的患者与NOTCH2基因(1p12)突变有关,与经典临床特征不完全符合,称之为Alagille综合征2型。
典型病例:女性,21岁,间断肝功异常21年,ALT/AST 40-300 U/L,ALP 100-400 U/L,GGT>1000 U/L,TBIL 17-40μmol/L。基因检测示JAG1基因exon13 c.C1653A p.C551X杂合突变(Likely Pathogenic)。
贮积/沉积型包括溶酶体贮积症、内质网贮积症、胞浆内贮积、色素沉积、铜铁沉积、卟啉结晶等。
戈谢病表现为条纹状胞浆,Kupffer细胞中CD68阳性,DPAS阳性;尼曼-匹克病表现为泡沫样胞浆,肝细胞及Kupffer细胞中CD68阳性,DPAS阴性;胆固醇酯贮积症表现为Kupffer细胞内泡沫样胞浆,含针状胆固醇结晶,DPAS阳性;胱氨酸贮积症表现为Kupffer细胞内胱氨酸结晶,偏振光下呈亮银色。
图4. 溶酶体贮积症导致的不同疾病的组织学特征
内质网贮积症
代表性的疾病有α1-抗胰蛋白酶缺乏症、α1-抗糜蛋白酶缺乏症、无/低纤维蛋白原血症、抗凝血酶Ⅲ缺乏症,其中,α1-抗胰蛋白酶缺乏症和无/低纤维蛋白原血症较常见。二者均可见胞质内嗜酸性小球,α1-抗胰蛋白酶缺乏症的小球分布于汇管区或间隔周围肝细胞内,呈D-PAS染色阳性,免疫组化染色显示α1-抗胰蛋白酶阳性;无/低纤维蛋白原血症的小球分布无区域性,D-PAS染色阴性或弱阳性,PTAH染色呈蓝紫色,免疫组化染色显示纤维蛋白原阳性。
图5. 内质网贮积症的组织学特征
胞浆内贮积包括糖原贮积症(尤其Ⅲ/Ⅳ型可进展为肝硬化)和Lafora病(肌阵挛性癫痫,EPM2A/NHLRC1基因突变)。
图6. 色素及结晶沉积的组织学特征
实例:男性,28岁,肝功能异常8年余,高脂血症,脂肪肝。2022年ALT 347 U/L,AST 116 U/L,ALP 76 U/L,GGT 46 U/L,ALB 20 g/L;2025年ALT 40 U/L,AST 58 U/L,ALP 38 U/L,GGT 94 U/L,ALB 45 g/L。病理检查示肝细胞内、巨噬细胞内胆固醇结晶,肉芽肿形成;汇管区内多核巨细胞伴胆固醇结晶。基因检测示CYP4V2基因复合杂合突变,诊断为Bietti结晶样视网膜营养不良,该病与脂肪酸代谢异常相关。
遗传代谢性肝病中表现为肝脏炎症坏死的常见疾病是α1-抗胰蛋白酶缺乏症和Wilson病。其中,Wilson病可表现为肝组织大块或亚大块坏死,需要与其他原因所致的肝衰竭进行鉴别,如药物性肝坏死、急性重症自身免疫性肝炎等。
图7. Wilson病的组织学特征
门静脉高压型主要包括HLA-DR3阳性、端粒酶缺陷综合征(TERT/TERC)、Turner综合征、Adams-Oliver综合征(NOTCH1)、CTC突变、FOPV突变等。
如一位20岁女性,由TERT基因突变所致短端粒综合征,其肝脏病理表现为汇管区纤维化伴细纤维隔形成,稍大的汇管区内门静脉分支管壁增厚,有的汇管区内未见相应口径的门脉支,肝实质内见肝细胞板萎缩带。病理诊断符合特发性非硬化性门脉高压(现称门静脉-肝窦血管病,PSVD)。
大部分遗传代谢性肝病具有一定的组织学特点,肝穿刺活检有助于明确诊断或提供重要信息。如先天性肝纤维化、Dubin-Johnson综合征、原卟啉病等具有特征性表现。根据病理特征,遗传代谢性肝病可分为肝组织基本正常型、脂肪变性型、胆汁淤积型、贮积/沉积型、肝炎型和门静脉高压型六大类型,各型可单独出现,也可合并存在。遗传代谢性肝病的表现复杂多样,需结合临床、病理、实验室检查及基因分析做出综合诊断。病理医师需要及时查阅最新文献,了解临床及基础研究进展,开展新技术,不断提高诊断水平。
参考文献
1.刘晖, 梁晨, 郑素军, 王泰龄. 遗传代谢性肝病的病理诊断思路. 中华肝脏病杂志, 2022,30(11): 1253-1258.
2.中国罕见病联盟遗传性肝病分会,中华医学会儿科学分会感染学组,中华儿科杂志编辑委员会. 家族性肝内胆汁淤积症诊疗专家共识(2026)[J]. 中华儿科杂志,2026,64(03):262-269.
刘晖 教授
首都医科大学附属北京佑安医院
首都医科大学附属北京佑安医院病理科
医学博士,主任医师
中华医学会肝病学分会遗传代谢性肝病协作组委员
中国研究型医院学会肝病学分会常务委员
全国疑难及重症肝病攻关协作组委员
北京医学会病理学分会妇儿学组委员
北京肿瘤病理精准诊断研究会肝胆胰分会常务委员
北京整合医学会肝脏临床病理学专委会常务委员
中华志愿者协会中西医结合专家志愿者委员会肝病科专业组常务委员
北京中西医结合学会精准医学专委会委员
长期从事肝脏病理研究,专业特长:病毒性肝炎、药物性肝损伤、自身免疫性肝病、胆管病、遗传代谢性肝病及肝脏肿瘤的病理诊断。
(来源:《国际肝病》编辑部)
声明:本文仅供医疗卫生专业人士了解最新医药资讯参考使用,不代表本平台观点。该信息不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议,如果该信息被用于资讯以外的目的,本站及作者不承担相关责任。